Número Atual: Julho-Setembro 2020 - Volume 4 - Número 3
- Imprimir
- Indicar
- Estatísticas
- (0)
Comentários - Como Citar
- Download da Citação
- Artigos Relacionados
-
Outros dos
Autores

Comunicação Clínica e Experimental
Nova mutação no gene STAT1 associada com candidíase mucocutânea crônica
New STAT1 gene mutation associated with chronic mucocutaneous candidiasis
Mariana Jobim1; Anne Puel2; Gisele Ewald1; Beatriz Chamun Gil1; Melanie Migaud2; Iara Santos Fagundes1; Jaqueline Cardone1; Luiz Jobim1,3
DOI: 10.5935/2526-5393.20200054
1. Hospital de Clínicas de Porto Alegre, Serviço de Alergia e Imunologia - Porto Alegre, RS, Brasi
2. Universidade de Paris, Laboratório de Genética Humana de Doenças Infecciosas - Paris, Ilha de França, França
3. Universidade Federal do Rio Grande do Sul, Departamento de Medicina Interna - Porto Alegre, RS, Brasil
Endereço para correspondência:
Luiz Jobim
E-mail: ljobim@hcpa.edu.br
Submetido em: 09/09/2020
Aceito em: 12/09/2020
Não foram declarados conflitos de interesse associados à publicação deste artigo
RESUMO
Mutações no gene STAT1 (signal transducer and activator of transcription 1) têm sido identificadas como responsáveis pela maioria dos casos sindrômicos da candidíase mucocutânea crônica com herança autossômica dominante (AD). Nesse artigo, descrevemos uma menina de 7 anos que apresentou candidíase da mucosa oral e unhas, além de infecção disseminada da pele e couro cabeludo por Microspora gipseum. Recentemente, a paciente foi diagnosticada e tratada de meningite por Cryptococcus neoformans. Na família não existem outros casos de candidíase. A avaliação imunológica incluiu a detecção de subpopulações de linfócitos (CD3, CD4, CD8, CD20 e células NK), assim como a dosagem de IgG, IgA, IgM e IgE, subclasses de IgG e autoanticorpos. Excluindo-se discreta diminuição de CD3, CD4, CD8, NK e leve aumento de IgG1, os demais exames estiveram dentro da normalidade. O sequenciamento do exoma detectou uma rara mutação em heterozigose no exon 14 do domínio de ligação do DNA (DNA-binding domain) do gene STAT1, ocasionando um provável ganho de função (GOF) responsável pela doença (Gly384Asp). Essa variação foi também identificada pelo sequenciamento de Sanger, não estando reportada nos bancos de dados públicos e apresentando elevado potencial de dano (índice CADD=32). Será interessante contarmos com informações clínicas e estudos com outros pacientes para conhecermos mais essa mutação patológica. Além da apresentação do caso, discutiremos as formas de tratamento existentes.
Descritores: Candidíase mucocutânea crônica, fator de transcrição STAT1, interleucina-17, células Th17.
INTRODUÇÃO
A candidíase mucocutânea (CM) é caracterizada por infecções por Candida sp., afetando as unhas, pele, couro cabeludo, mucosa oral e genital. A candidíase das mucosas é comum em indivíduos tratados com antibióticos e corticosteroides. A maioria das mulheres apresenta pelo menos um episódio de candidíase vaginal durante a vida.
A candidíase mucocutânea crônica (CMC) é uma doença caracterizada pela infecção crônica e persistente da pele, unhas e mucosas. Pode estar associada com imunodeficiências que afetam a imunidade celular como acontece na AIDS e na imunodeficiência severa combinada em suas diversas formas, sendo classificada como CMC doença1.
A CMC sindrômica (CMCS) ocorre quando está associada com outras manifestações clínicas infecciosas e autoimunes. Nesses pacientes existe um distúrbio da imunidade mediada pela interleucina 17 (IL-17), citocina reconhecida como responsável pela defesa contra a cândida e outros fungos, como o Microsporum sp. A neutralização da IL-17 com anticorpos monoclonais usados para tratamento da psoríase tem como efeito colateral a possibilidade de desencadear candidíase. Mutações em diversos genes podem interferir na imunidade mediada pela IL-17, podendo ser encontradas também em imunodeficiências primárias sem aparente distúrbio da imunidade celular1,2.
A imunodeficiência com mutação AD com ganho de função (GOF) no gene STAT1 (Signal Transducer and Activator of Transcription-1) é a mais frequente doença genética responsável por CMC sindrômica, relacionada com disfunção da imunidade atribuída às células Th17 produtoras de IL-173-5. Os pacientes apresentam também outras infecções mucocutâneas bacterianas, virais e fúngicas, assim como predisposição à autoimunidade. Nesses pacientes observase aumento da fosforilação do STAT1 e deficiência da desfosforilação nuclear, levando à diminuição da transcrição que afeta a liberação de IL-17A e IL-17F e outras citocinas4.
Na última década foram descritos muitos pacientes com mutação do tipo GOF no gene STAT1, sendo que um estudo internacional analisou 274 pacientes de 167 famílias. As mutações ocorrem principalmente no domínio de dupla espiral (doble coiled-coin domain) em 62% dos pacientes e no domínio de ligação com o DNA (DNA-binding domain) em 35%. A média de idade foi de 22 anos, sendo que 98% dos pacientes apresentavam CMC. Frequentemente tiveram infecções por Staphylococcus aureus na pele e aparelho respiratório (36%), assim como infecções virais, na maioria, herpética (38%). Alguns pacientes foram acometidos por autoimunidade (37%), como hipotireoidismo (22%) e diabete tipo I (4%). Aneurisma cerebral (6%) e câncer (6%) foram as comorbidades de pior prognóstico6.
Outra doença responsável por quadro sindrômico de CMC é a imunodeficiência primária autossômica recessiva (AR), conhecida por síndrome de Hiper-IgE, e que acontece pela mutação no gene STAT3. Nessa patologia existe diminuição importante de células Th17 e da IL-17A e IL-22 decorrente da sinalização inadequada de STAT3. Os pacientes são suscetíveis à CMC, infecções da pele e pulmão com estafilococo, além de eczema, IgE elevada, retenção dentária e face característica.
Pacientes com deficiência AR dos genes da proteína p40 e do receptor de IL-12 (IL-12p40 e IL12Rb1) podem ter susceptibilidade às micobactérias e salmonelas7. Ocasionalmente desenvolvem CMC sindrômica por diminuição de IL-17A e INF-g.
Pacientes com a síndrome Poliendocrinopatia Autoimune com Candidíase e Distrofia Ectodérmica (APECED) apresentam-se na infância com a combinação de candidíase, hipoparatireoidismo, hipogonadismo e insuficiência suprarrenal autoimune. Eles também apresentam alopécia areata, vitiligo e distrofia ectodérmica. A doença é causada por mutações AR no gene AIRE (21q22.3) que codifica para o fator de transcrição envolvido em mecanismos de tolerância imunológica que contribui para a seleção negativa de linfócitos autor-reativos no timo, linfonodos e baço. Um distúrbio imunológico faz com que exista a produção de anticorpos contra a IL-17A, IL-17F e ou IL-22. A autoimunidade é responsável pelo ataque aos órgãos endócrinos, assim como contra a imunidade mediada pela IL-172.
Pacientes com deficiência AR de CARD9 (caspase recruitment domain-containing protein 9 ) desenvolvem infecções com micobactérias e também CMC sindrômica e invasiva, associada com dermatofitose. Raros pacientes foram identificados com esta síndrome8. A CARD9 é uma proteína de sinalização com papel importante na imunidade inata e adaptativa, permitindo uma cascata de citocinas inflamatórias responsáveis pela defesa contra fungos.
Definiu-se como CMC doença (CMCD) a patologia que afeta os pacientes na ausência de outros sinais clínicos proeminentes, referindo-se aos pacientes com CMC como fenótipo clínico principal. A etiologia não está relacionada com genes que causam imunodeficiência severa combinada ou com outras imunodeficiências, nem com os genes responsáveis pela CMC sindrômica. Quatro tipos de alterações genéticas existem associados com esse fenótipo. Deficiência AR do receptor de IL-17A, IL-17C e do ACT1, além da deficiência AD de IL-17F. Cada um desses defeitos tem impacto na sinalização de IL-17, com diminuição da imunidade à cândida e CMCD2.
DESCRIÇÃO DO CASO
Paciente com 7 anos, sexo feminino, apresentou candidíase nas unhas e mucosa oral desde o primeiro ano de idade. A paciente consultou em diversas especialidades. Na Pediatria, acompanhou desnutrição e infecções bronco-pulmonares. Na Otorrino, tratou otite média serosa com eventual colocação de tubos de aeração. Recentemente foi identificada a presença do ácaro Demodex folliculorum na secreção do ouvido externo. Na Dermatologia diagnosticaram CMC com todas as unhas afetadas e dermatofitose extensa (Microspora gypseum), atingindo a pele e couro cabeludo, tendo sido internada por essa causa.
Um quadro clínico grave aconteceu posteriormente, sendo internada com cefaleia intensa, febre, bradicardia e rebaixamento do sensório. Além disso, apresentou tosse, secreção no ouvido direito, desnutrição e onicomicose. Foi diagnosticada meningite por Cryptococcus sp., tendo sido tratada com fluconazol, anfotericina B e flucitosina, além de ciprofloxacino. O Rx de tórax mostrou infiltrado pulmonar, adenopatias e escavações no lobo superior direito. Na tomografia de tórax observou-se aspecto de vidro fosco e escavações. Pesquisa direta, teste de látex e cultura no líquor foram positivos para Cryptococcus neoformans.
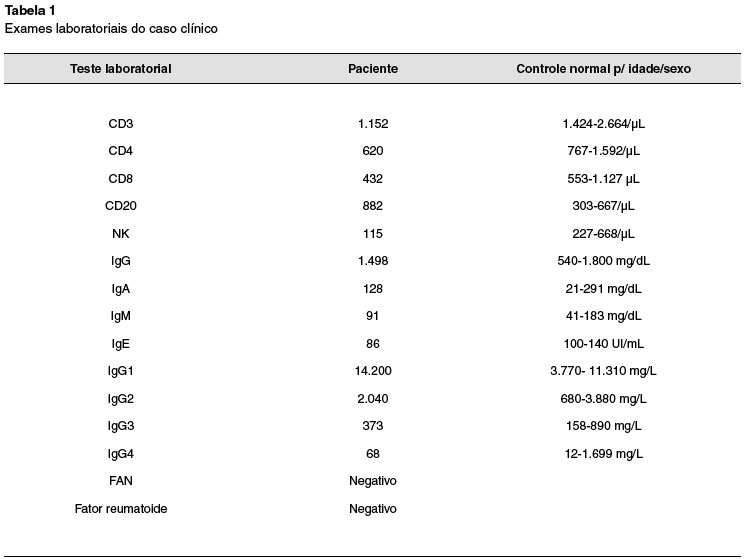
Do ponto de vista imunológico, a paciente apresentou discreta linfopenia (CD3, CD4, CD8 e NK), imunoglobulinas normais e leve aumento de subclasse de IgG1. As demais subclasses foram normais, assim como ausência de autoanticorpos (Tabela 1). Em relação à linfopenia, os achados não têm explicação adequada, mesmo por terem sido flutuantes e não refletirem uma definitiva citopenia, entretanto, a paciente sofre de uma clara deficiência do seu sistema imune em relação às defesas contra infecções fúngicas. A paciente recuperou-se sem sequelas da meningite, estando em acompanhamento ambulatorial.
A CMC foi considerada sindrômica pela diversidade de infecções, assim como pela identificação por sequenciamento do exoma de uma rara mutação GOF no exon 14 do domínio de ligação com o DNA do gene STAT1 (Gly384Val), causando a doença (Figura 1). Essa mutação, em heterozigose, ainda não estava reportada nos bancos de dados internacionais, entretanto, outras mutações patogênicas semelhantes já foram publicadas (Gly384Asp e Gly384Cys).
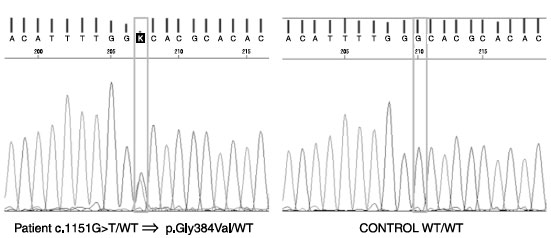
Figura 1
Sequenciamento do exon 14 do domínio de ligação com o DNA do gene STAT1. Observa-se rara mutação GOF em heterozigose (Gly384Val) em relação ao DNA controle
Foi calculado o valor do CADD (combined annotation dependent depletion), sendo uma ferramenta útil para predizer a gravidade das variantes/mutações de nucleotídeos encontradas num indivíduo. Para o valor encontrado de 32, considerase ter um alto potencial de dano9.
DISCUSSÃO
O tratamento de pacientes com mutações em STAT1 tem sido realizado com medicações antibacterianas, antivirais, antifúngicas e eventualmente com imunoglobulina endovenosa e imunossupressores. A problemática que envolve esses pacientes é decorrente da falta de terapêutica efetiva na redução ou cura da infecção crônica e ou da autoimunidade. Existem duas tentativas de intervenção, além do tratamento sintomático, devendo-se citar o uso de ruxolitinib, droga inibidora de JAK1/2 usada no tratamento da mielofibrose, e o transplante de células-tronco hematopoiéticas (TCTH). O uso de imunossupressão para tratar casos associados com autoimunidade tem demonstrado piorar os processos infecciosos.
O tratamento de uma criança de 12 anos de idade com mutação GOF em STAT1 e CMCS recalcitrante demonstrou ser clinicamente reversível in vitro e in vivo com ruxolitinib. A melhora clínica foi alcançada após 8 semanas de tratamento, via oral, quando foi conseguida a supressão da fosforilação de STAT1, STAT3 e STAT5 induzida pelo IFN-g e IFN-α. A piora clínica aconteceu concomitantemente com o aumento da atividade de fosforilação de STAT1, após a retirada da medicação, voltando ao normal com a reintrodução10.
Um paciente de 10 anos com CMCS e citopenia autoimune foi tratado com ruxolitinib, observandose aumento da fosforilação de STAT1, sem afetar a cinética da desfosforilação. Foi observado que o paciente normalizou a diferenciação de linfócitos Th17, curou a candidíase e obteve remissão da citopenia11.
Uma criança de 3 anos de idade com a mutação GOF do STAT1 (1154C>T, Thr385Met), presença de severa autoimunidade (citopenia, hepatite autoimune e disfunção renal) e com susceptibilidade a infecções (cândida e CMV), foi tratada com ruxolitinib. O nível de IL-17 não aumentou, mas o INF-g diminuiu após o quarto mês de tratamento. A fosforilação de STAT1 diminuiu significantemente após estimulação das células com INF-g. O paciente estava no sexto mês de ruxolitinib quando publicaram a experiência, tendo melhorado da autoimunidade e controlada a infecção por CMV e candidíase12.
Os riscos do uso do ruxolitinib foram sinalizados, após a análise de dois pacientes adultos com mutações no STAT1. Eles apresentavam infecções importantes, o primeiro com Trichophiton interdigitale e Trichophiton mentagrophite, e o segundo com coccidioidomicose. Ambos foram tratados com 20 mg de 12 em 12 horas da medicação oral e foram analisados in vitro para observação dos efeitos. Nenhum dos dois pacientes melhorou, embora tenham inibido a fosforilação ex vivo e in vitro de STAT1. Não foi observado aumento do número de linfócitos Th17 e da produção de IL-17. Com isso, o estudo concluiu que a dose do medicamento para alcançar os efeitos clínicos desejados deve ser maior do que a dose aprovada para uso, pelo menos para os fungos que os pacientes apresentavam13. As publicações que mostram efeito benéfico em pacientes com a mutação GOF no STAT1 foram todas com CMC e não com outros fungos invasivos.
O transplante de células-tronco hematopoiéticas (TCTH) tem sido usado em pacientes com sintomas severos decorrentes de mutações em STAT1 (GOF), entretanto, esse tratamento não está bem estabelecido devido a alguns resultados insatisfatórios. Um estudo com 15 pacientes de uma coorte internacional foi publicado14. Todos os pacientes tinham mutações em heterozigose, tanto no domínio de dupla espiral (doble coiled-coin domain) como no domínio de ligação com o DNA (DNA-binding domain). O enxerto foi aceito em 75% dos casos, entretanto, a sobrevida foi somente de 40%. Seis pacientes sobreviveram ao primeiro ano do transplante, e nove faleceram. Entre os sobreviventes, cinco tiveram reconstituição completa e um apresentou quadro de quimera mista. O intervalo sem doença após o transplante foi pequeno. Infecções ocorreram em todos os pacientes antes do transplante, especialmente por fungos. Doze pacientes apresentaram CMC nas unhas, pele, mucosa oral e trato gastrointestinal. Cinco pacientes apresentaram infecções fúngicas invasivas, incluindo aspergilose pulmonar, candidemia e meningite por criptococos. Infecções pulmonares foram frequentes, com bactérias que inclusive causaram septicemia. Outros vírus também foram observados como varicela zoster, CMV, EBV, BK vírus, parvovírus B19 e herpes simples tipo 6. Infecções com micobactérias foram identificadas em três pacientes. Autoimunidade foi observada em 10 pacientes que apresentaram síndrome semelhante a IPEX (imunodeficiência, poliendocrinopatia e enteropatia ligada ao X), neutropenia, anemia hemolítica, tireoidite e diabete. Histiocitose hemofagocítica (HLH) aconteceu em dois pacientes e a deficiência de anticorpos foi observada em nove, tendo sido tratadas com imunoglobulina EV. Defeitos da imunidade celular e humoral ocorreram independentes da presença de autoimunidade. A conclusão dos autores foi de que o TCTH em pacientes com mutações GOF no gene STAT1 pode ocasionar a cura do paciente, mas as complicações associadas podem levar à falha secundária do enxerto e diminuição da sobrevida. A crítica que se pode fazer ao trabalho é de que os pacientes foram tratados por equipes e protocolos diversos, prejudicando a análise dos resultados. Os transplantes, nos casos mais graves, talvez devam ser realizados precocemente.
Dois pacientes foram transplantados recentemente, sendo que o primeiro com sangue de cordão umbilical de gêmeo HLA idêntico, e o segundo recebeu a medula óssea de doador não relacionado, mas com compatibilidade total no sistema HLA. O paciente que recebeu o sangue de cordão recuperou-se totalmente, entretanto o outro apresentou infecção sistêmica com CMV, falha do enxerto e hemorragia pulmonar fatal. A desregulação da produção de IFN-g, a supressão da resposta de IL-17 e o aumento da fosforilação de STAT1 foram normalizados após o transplante no primeiro paciente. A conclusão é que o TCTH pode ser uma alternativa de cura para os pacientes com doença progressiva que não respondem adequadamente ao tratamento convencional.
No caso do paciente relatado não existe doador na família, sendo necessária a identificação no REDOME (Registro de Doadores de Medula Óssea) para um transplante com doador adequado. No entanto, desistimos de indicar o transplante devido aos resultados inconsistentes da literatura. O tratamento com a medicação oral ruxolitinib é extremamente dispendioso, estando o paciente esperando resultado de solicitação oficial.
Este caso clínico chama a atenção para os pacientes com infecções fúngicas graves, associadas com infecções bacterianas e autoimunidade. Esses pacientes devem ser avaliados genética e imunologicamente para as diversas possibilidades de defeitos na imunidade decorrentes da IL-17, citocina responsável pela CMC sindrômica.
REFERÊNCIAS
1. Puel A. Human inborn errors of immunity underlying superficial or invasive candidiasis. Hum Genet. 2020 Jun;139(6-7):1011-22.
2. Okada S, Puel A, Casanova JL, Kobayashi M. Chronic mucocutaneous candidiasis disease associated with inborn errors of IL-17 immunity. Clin Transl Immunology. 2016 Dec 2;5(12):e114. doi: 10.1038/cti.2016.71.
3. Van De Veerdonk FL, Plantinga TS, Hoischen A, Smeekens SP, Joosten LAB, Gilissen C, et al. STAT1 mutations in autosomal dominant chronic mucocutaneous candidiasis. N Engl J Med. 2011;365(1):54-61.
4. Liu L, Okada S, Kong XF, Kreins AY, Cypowyj S, Abhyankar A, et al. Gain-of-function human STAT1 mutations impair IL-17 immunity and underlie chronic mucocutaneous candidiasis. J Exp Med. 2011;208(18):1635-48.
5. Puel A, Cypowyj S, Maródi L, Abel L, Picard C, Casanova JL. Inborn errors of human IL-17 immunity underlie chronic mucocutaneous candidiasis. Curr Opin Allergy Clin Immunol. 2012;12(6):616-22.
6. Toubiana J, Okada S, Hiller J, Oleastro M, Gomez ML, Becerra JCA, et al. Heterozygous STAT1 gain-of-function mutations underlie an unexpectedly broad clinical phenotype. Blood. 2016;127(25):3154-64.
7. Al-Muhsen S, Casanova JL. The genetic heterogeneity of mendelian susceptibility to mycobacterial diseases. J Allergy Clin Immunol. 2008;122(6):1043-51.
8. Grumach AS, de Queiroz-Telles F, Migaud M, Lanternier F, Filho NR, Palma SMU, et al. A Homozygous CARD9 Mutation in a Brazilian Patient with Deep Dermatophytosis. J Clin Immunol. 2015;35(5):486-90.
9. Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019;47(1):886-94.
10. Bloomfield M, Kanderová V, Para ková Z, Vrabcová P, Svato M, Froková E, et al. Utility of ruxolitinib in a child with chronic mucocutaneous candidiasis caused by a novel STAT1 gain-offunction mutation. J Clin Immunol. 2018;38(5):589-601.
11. Weinacht KG, Charbonnier LM, Alroqi F, Plant A, Qiao Q, Wu H, et al. Ruxolitinib reverses dysregulated T helper cell responses and controls autoimmunity caused by a novel signal transducer and activator of transcription 1 (STAT1) gain-of-function mutation. J Allergy Clin Immunol. 2017;139(5):1629-40.
12. Moriya K, Suzuki T, Uchida N, Nakano T, Katayama S, Irie M, et al. Ruxolitinib treatment of a patient with steroid-dependent severe autoimmunity due to STAT1 gain-of-function mutation. Int J Hematol. 2020;112(2):258-62. doi: 10.1007/s12185-020-02860-7.
13. Zimmerman O, Rösler B, Zerbe CS, Rosen LB, Hsu AP, Uzel G, et al. Risks of Ruxolitinib in STAT1 Gain-of-Function-Associated Severe Fungal Disease. Open Forum Infect Dis. 2017;4(4):ofx202. doi: 10.1093/ofid/ofx202.
14. Leiding JW, Okada S, Hagin D, Abinun M, Shcherbina A, Balashov DN, et al. Hematopoietic stem cell transplantation in patients with gain-of-function signal transducer and activator of transcription 1 mutations. J Allergy Clin Immunol. 2018;141(2):704-17.
2025 Associação Brasileira de Alergia e Imunologia
Rua Domingos de Morais, 2187 - 3° andar - Salas 315-317 - Vila Mariana - CEP 04035-000 - São Paulo, SP - Brasil - Fone: (11) 5575.6888